
发布日期: 2025-04-27 12:21:01
在日常工作或社交场景中,屏幕截图已成为高频操作。但许多人并未意识到,截图文件...

发布日期: 2025-04-26 12:26:38
CSV文件作为数据处理领域的通用载体,其结构管理直接影响着工作效率。对于需要频繁...

发布日期: 2025-04-13 11:41:58
在互联网深度融入日常生活的当下,浏览器存储的网站数据逐渐成为隐私泄露的高危区...
发布日期: 2025-04-29 17:30:31
数字化浪潮席卷全球博物馆行业,数据管理效率成为衡量机构现代化水平的重要指标。...

发布日期: 2025-05-20 18:36:20
在工业监测、环境研究及医疗健康等领域,连续、精准的数据采集与标记能力直接影响...
发布日期: 2025-03-28 09:00:01
上海某私募基金交易员李明习惯在开盘前打开一款名为"MarketPulse Pro"的股票客户端。这...

发布日期: 2025-04-19 13:43:14
在数据中心运维与数字取证领域,某款支持多磁盘并行分析的专业工具近期引发行业关...

发布日期: 2025-04-20 16:34:36
微信公众号运营过程中,数据监测与分析是内容迭代的重要环节。面对后台海量的图文...

发布日期: 2025-05-05 19:12:28
在信息爆炸的互联网时代,一个吸睛的社交媒体账号名称如同数字世界的黄金广告位。...
发布日期: 2025-05-07 17:49:06
在信息爆炸的时代,数据决策成为企业发展的核心能力。原始表格数据往往如同一座迷...

发布日期: 2025-03-26 16:17:00
在信息爆炸的时代,RSS订阅依然是许多人高效获取内容的核心工具。相较于臃肿的网页...

发布日期: 2025-05-12 15:11:33
在数据库开发过程中,存储过程的调试长期困扰着开发者。传统的手动调试方式不仅效...
发布日期: 2025-05-09 13:36:22
数据采集领域长期存在一个痛点:爬虫抓取的海量信息如何快速整理成可读、可分析的...

发布日期: 2025-05-09 11:16:59
在信息爆炸的互联网时代,快速获取网页源码的需求日益增长。无论是开发者调试代码...

发布日期: 2025-05-21 17:35:14
在工业自动化、物流仓储及实验室检测领域,电子秤数据的高频采集直接影响着作业效...

发布日期: 2025-05-12 11:24:34
宿舍角落里堆积的旧教材、衣柜深处不再合身的连衣裙、闲置多年的二手自行车……每...
发布日期: 2025-05-05 19:05:17
在数据中心规模持续扩张的当下,某科技团队研发的NetHealth评分系统已在国内15个省级...

发布日期: 2025-05-02 19:37:10
在数据驱动的商业环境中,自动化采集工具已成为企业获取市场情报的刚需。近期业内...

发布日期: 2025-05-12 12:43:46
在信息爆炸的时代,网页表格作为数据存储的重要载体,广泛存在于企业报表、学术研...

发布日期: 2025-05-27 10:26:47
在数据分析领域,80%的时间被消耗在数据预处理环节。面对动辄百万行的CSV数据集,传...
发布日期: 2025-05-24 14:35:17
在数据处理成为日常工作的今天,跨平台工具的需求日益增长。某技术团队近期推出的...

发布日期: 2025-04-18 09:19:05
音视频元数据编辑工具:ID3标签修改指南 在数字媒体时代,音乐和音频文件的管理逐渐...

发布日期: 2025-05-22 16:21:42
在信息爆炸的时代,桌面便签成为许多人记录待办事项、临时灵感的核心工具。但随着...
发布日期: 2025-04-23 10:32:09
电子表格数据清洗转换器作为数据处理领域的实用工具,正逐渐成为企业及个人用户优...
发布日期: 2025-05-09 18:08:59
Cookie作为用户身份识别与状态维持的重要数据载体,在网页开发、数据分析、自动化测...

发布日期: 2025-04-04 11:19:04
数据安全传输与一致性维护是数字资产管理的核心命题。面对跨地域服务器同步、分布...
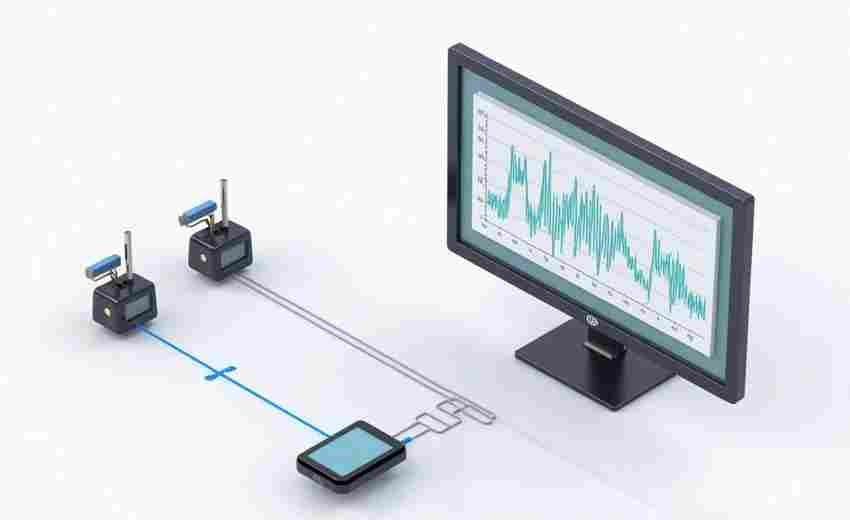
发布日期: 2025-03-21 12:00:01
在精密实验场景中,温度每偏差1℃可能改变化学反应速率,湿度波动3%会导致精密仪器...

发布日期: 2025-05-04 14:57:56
在数字影像管理领域,EXIF信息如同胶卷时代的拍摄笔记,记录着每张照片的技术基因。...
发布日期: 2025-04-30 13:01:55
随着工业物联网与智慧农业的快速发展,环境数据的采集与分析需求呈现爆发式增长。...

发布日期: 2025-05-13 12:31:11
在数据驱动决策的时代,网页抓取技术已成为企业获取商业情报的重要手段。面对市场...

发布日期: 2025-03-23 09:57:00
在日常办公场景中,Excel表格承载着大量关键数据,但人工校对不仅耗时,且易因重复...

发布日期: 2025-04-03 11:31:47
在数据交换需求频繁的办公场景中,FTP协议仍是跨平台传输的可靠选择。Python生态圈提...

发布日期: 2025-04-13 18:13:21
在信息泄露频发的时代,密码如同个人资产的最后一道屏障。随机密码生成器作为数字...

发布日期: 2025-05-23 10:50:28
数字时代,数据安全成为企业生存的底线。网络关键词作为品牌运营、用户洞察的核心...

发布日期: 2025-05-03 09:38:42
互联网时代,信息过载催生了书签管理的刚性需求。当个人收藏链接突破四位数时,混...

发布日期: 2025-04-20 15:37:44
随着老龄化社会加速,养老机构床位的供需矛盾日益突出。传统模式下,家属需通过电...
发布日期: 2025-04-20 15:51:36
电子表格已成为现代办公场景中数据管理的核心载体,随着文件版本迭代频率的加速,...

发布日期: 2025-03-28 12:14:07
在信息爆炸的互联网时代,快速获取网页核心内容成为数据分析、舆情监测等领域的关...

发布日期: 2025-04-17 14:50:18
在信息爆炸的时代,微信已成为个人与企业的核心沟通工具。每天面对海量消息,如何...
发布日期: 2025-05-05 11:29:36
在快节奏的现代职场中,会议管理往往消耗大量时间。从预约参会人员到整理会议记录...

发布日期: 2025-04-13 19:43:21
足球比赛数据可视化工具近年来快速发展,其中雷达图对比功能成为业内关注焦点。该...

发布日期: 2025-05-17 11:25:54
在信息爆炸的社交媒体时代,微博作为国内最大的舆论场之一,实时捕捉热点关键词已...

发布日期: 2025-04-07 12:32:48
在信息爆炸的办公场景中,普通职场人日均接收的邮件数量从50封到200封不等。其中真...

发布日期: 2025-04-25 17:53:43
在财务数字化转型浪潮中,数据来源的复杂性呈指数级增长。一份报表的最终结果,可...
发布日期: 2025-05-06 09:00:02
工具定位与实际痛点 数据库运维与开发过程中,跨环境、跨版本的表结构同步一直是高...

发布日期: 2025-04-19 18:28:21
信息安全已成为数字生活的基础需求。一款基于PyQt5框架开发的本地化加密工具悄然流...

发布日期: 2025-04-07 12:57:33
当数码相机存储卡积累到第32GB时,摄影师王明发现他的工作流程出现了严重瓶颈——...
发布日期: 2025-05-07 15:39:57
在数字化时代,企业服务器和终端设备每时每刻产生海量网络流量数据。如何对这些动...

发布日期: 2025-04-03 18:54:01
在数字化办公场景中,频繁登录各类系统获取数据已成常态。某款基于Python开发的数据...

发布日期: 2025-04-11 15:15:00
在信息爆炸的互联网时代,快速获取有效内容成为刚需。无论是市场调研、竞品分析,...

发布日期: 2025-04-06 15:29:58
在数据驱动的业务场景中,快速获取并分析数据是企业决策的关键。传统数据库查询往...

发布日期: 2025-05-24 15:07:57
在数据驱动的商业环境中,数据库权限管理是保障企业信息安全的基石。随着业务复杂...
发布日期: 2025-05-19 18:25:49
在信息碎片化时代,数据呈现能力直接影响决策效率。一款零代码操作的可视化工具正...
发布日期: 2025-03-27 16:33:01
面对密密麻麻的销售记录、庞杂的财务数据或是海量的库存信息时,很多职场人都会陷...

发布日期: 2025-04-02 15:53:27
在南方梅雨季的清晨打开手机,屏幕左上角精确显示着"9点03分雨势减弱"的提示;北方...

发布日期: 2025-04-04 14:59:31
在数据量激增的办公场景中,Excel用户常面临一个痛点:如何快速识别并处理重复、近...

发布日期: 2025-03-26 17:10:20
在信息爆炸的时代,YouTube每天新增数百万条视频内容。无论是自媒体运营、学术研究还...

发布日期: 2025-05-16 14:16:16
在软件开发和测试环节中,真实数据的缺失常常成为效率瓶颈。无论是验证表单功能、...

发布日期: 2025-05-20 19:19:10
在数字信息爆炸的今天,文件管理已成为困扰许多职场人士的难题。某互联网公司的运...

发布日期: 2025-05-16 10:41:12
在金融、法律、医疗等专业领域,PDF文件中的表格承载着大量核心数据。某国际会计师...
